答疑解惑 | 华银康高通量测序检测中心带你解读转录组常见问题,洞悉数据分析内容!
2022-11-16 阅读数:9430
当我们拿到转录组测序报告和数据结果时,是否对报告中的数据内容有疑惑?怎么在文章中阐述这些数据所代表的意义?今天华银康高通量测序检测中心为大家整理了转录组常见问题解答内容,干货满满,快来一起学习!
Q1:转录组测序建库方式有哪些?该如何选择?
A1:原核生物样本采用的是去核糖体建库方式。由于原核生物中的mRNA上没有polyA尾,没法通过带有Oligo(dT)的磁珠富集。所以可以用试剂盒去除rRNA,再对所有的mRNA、非编码RNA(Non-coding RNA)以及游离的RNA进行富集。
真核生物可以采用去核糖体建库、Oligo(dT)富集mRNA这两种建库方式,由于真核生物mRNA的3’端存在polyA尾,可以用磁珠富集捕获到mRNA。
这两种建库方式需要看样本类型和RIN值(RNA完整性)来选择,比如RIN值在7以上可以采用Oligo(dT)建库方式。反之,可以进行去核糖体建库方式。由于Oligo(dT)只是富集到的mRNA的3’端,如果片段发生断裂会导致5’端的序列丢失,导致获得的mRNA完整性不足,所以这一建库方式对RNA完整性要求比较高。
Q2:转录组测序报告和数据结果中重要的内容有哪些?
A2:首先,我们用RSEM(RNASeq by Expectation Maximization) 工具进行基因以及转录本的表达定量,从而得到样本中所有表达的基因定量结果。随后,采用edgeR软件进行比较组中差异表达基因分析。最后,对这些筛选到的差异基因进行GO和KEGG功能富集分析,对功能通路进行注释以及分析通路上富集到的差异基因。
Q3:如何筛选差异基因?怎么看目标基因在实验组高/低表达?
A3:我们基于之前分析的所有转录本表达量数据文件,按照|log2FC|>= 1和 Pvalue < 0.05这两个阈值条件进行差异基因的筛选,那么FC的阈值范围为FC>=2 或者FC<=-2。如果log2FC大于0,就说明这个基因在实验组是高表达的,反之,log2FC小于0,说明在实验组是低表达的。
Q4:在基因差异表达分析中,例如样品1 vs 样品2,如何理解上调和下调?
A4:‘样品1 vs 样品2’,则样品1是对照组,样品2是处理组。在相应的结果文件1-vs-2.GeneDiffExp.xls和1-vs-2.GeneDiffExpFilter.xls中,如果一个基因被记为是上调,就表明相对于样品1(对照组),此基因在样品2(处理组)中的表达量是上调的。
Q5:GSEA富集分析哪些数据内容?样本数的要求?
A5:GSEA富集分析能帮助科研工作者们在两种不同的生物学状态 (biological states)中,判断某一组有特定意义的基因集合的表达模式更接近于哪一种。因此GSEA是一种非常常见且实用的分析方法,可以将数个基因组成的基因集与整个转录组、修饰组等做出简单而清晰的关联分析。
一般我们GSEA富集分析要求的样本数在3个生物学重复以上。
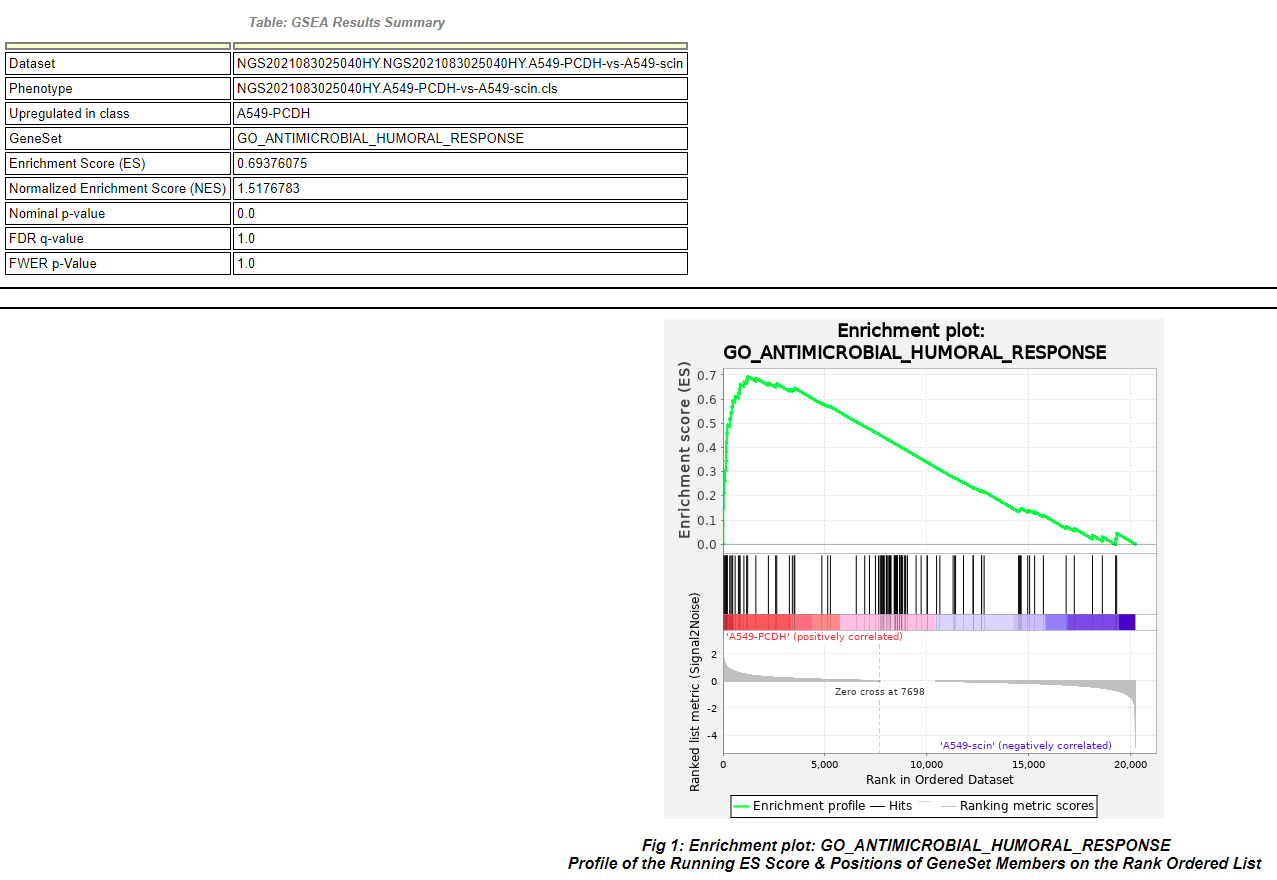
那么GSEA热图怎么看呢?
热图显示了前缘子集中的(聚类)基因。在热图中,表达值表示为颜色,其中颜色范围(红色,粉红色,浅蓝色,深蓝色)显示了表达值的范围(高,中,中,低,最低)。
对照组和处理组表达量高低的问题?
可以看热图中|ES|最大值对应的分组,就说明该基因集在这个分组中高表达,处于一个激活的状态,|ES|最高点右侧基因集属于核心基因集,对应表格中CORE ENRICHMENT为“Yes”的是核心基因集;
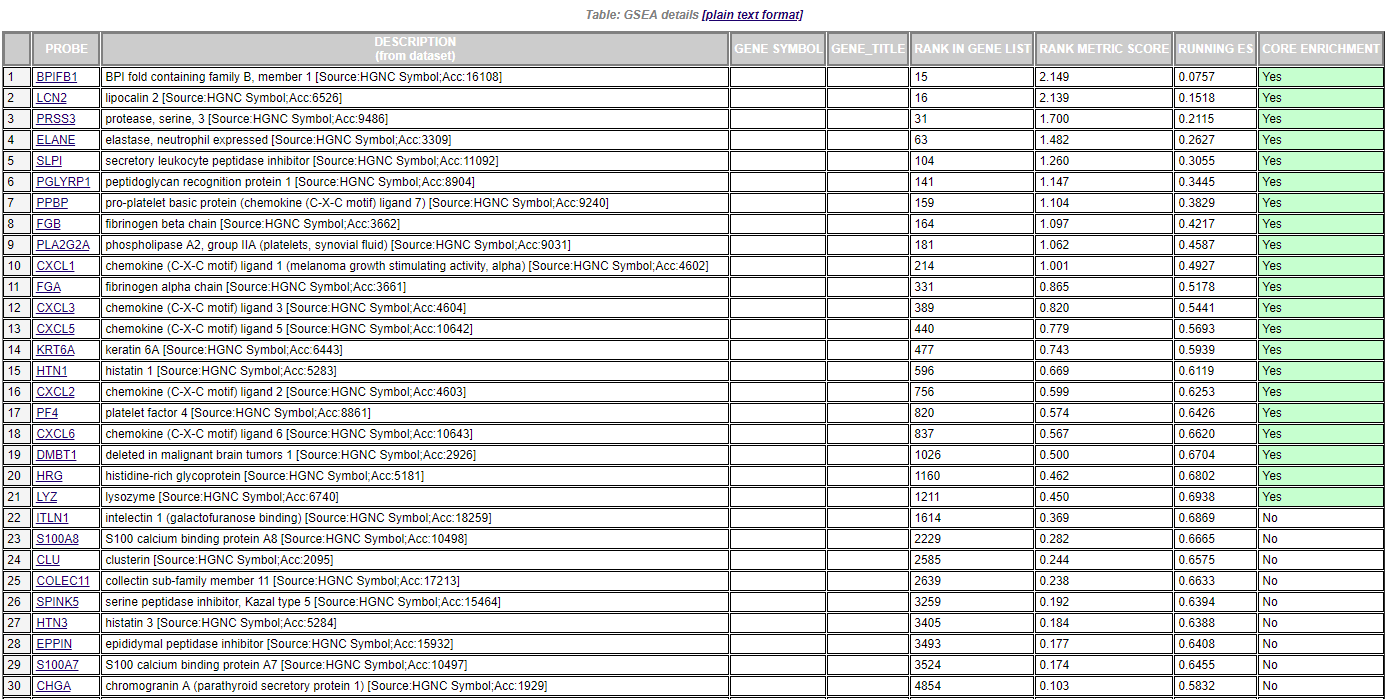
前面我们提到了ES,它的具体含义是什么呢?
每个基因对应的累计值就叫做富集得分 (Enrichment score, ES) ,而这个基因集的富集得分 (ES)则定义为遍历基因列表时遇到的离零的最大偏差,即峰值。峰值为正值表示基因集富集在列表的顶部(mut),负值表示富集在底部(wt)。
Q6:基因的表达量是基于什么计算的?
A6:表达定量的结果以FPKM为单位,具体计算公式如下。
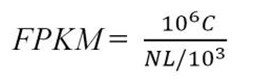
RPKM:Reads Per Kilobase Million,指的是每一百条reads中,对基因的每1000个base而言比对到的reads数。RPKM用于双端测序结果,由于每个fragment会包含两个reads,使用FPKM计算基因的表达量时,可以避免同一个fragment的两个reads计算2次的问题。
Q7:KEGG中level1、2、3、4层级代表的含义?
A7:KEGG是一个综合性公共数据库,首先,生物代谢通路主要分为6类,分别为:细胞过程(Cellular Processes)、环境信息处理(Environmental Information Processing)、遗传信息处理(Genetic Information Processing)、人类疾病(Human Diseases)、新陈代谢(Metabolism)、生物体系统(Organismal Systems),其中每类又被系统分类为二、三、四层。第二层又分为一些子pathway,第三层为其代谢通路图;第四层为每个代谢通路图的具体注释信息。
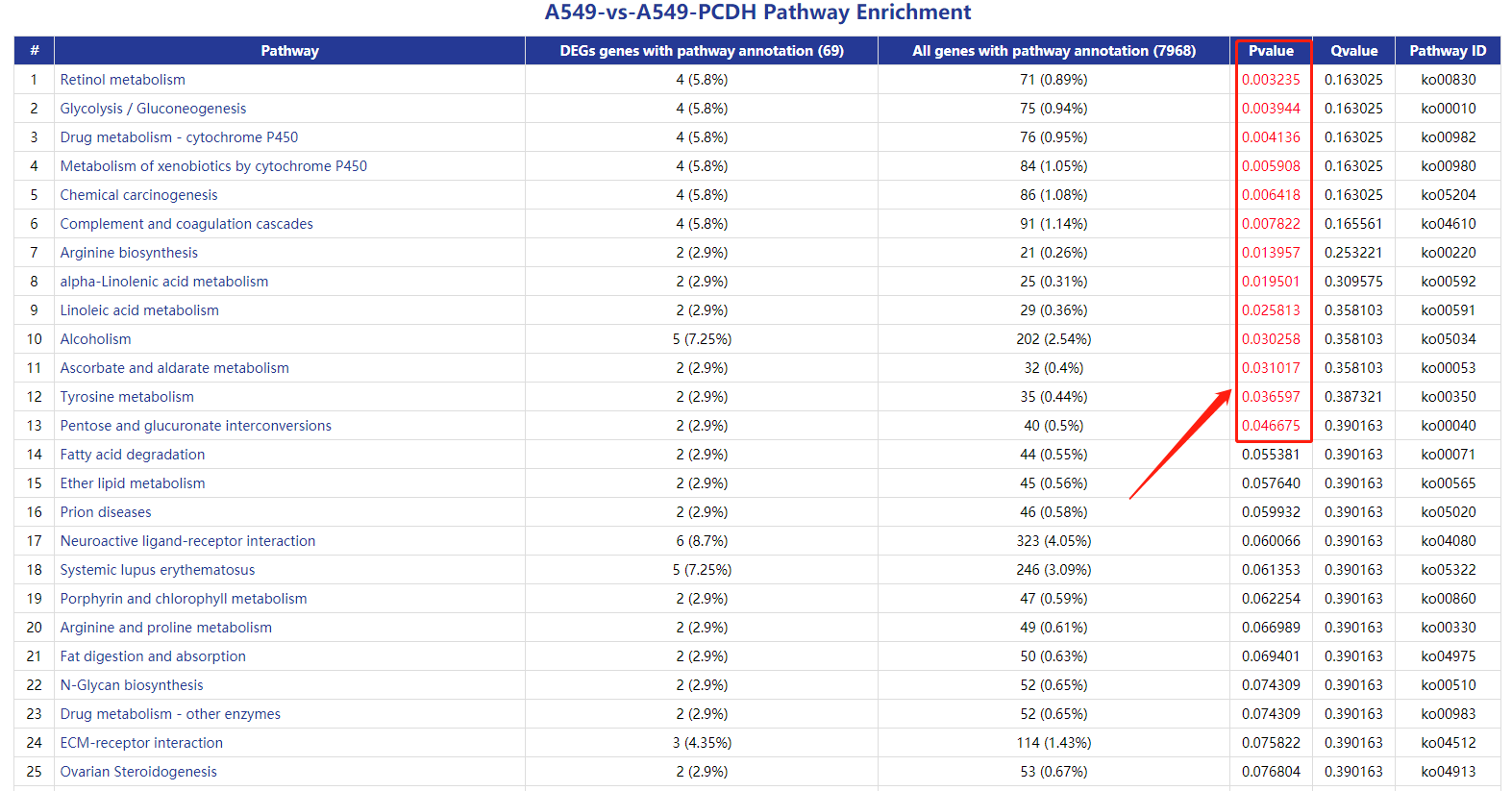
怎么看目标通路是否显著?怎么看通路上富集基因表达的上/下调?
p<0.05时,目标通路是显著的,通过KEGG代谢通路的map图可以看基因表达上/下调的情况,并且可以看出基因在通路上下游的位置,一般在前面的都处于上游的位置,并可以进行后续的基因敲除实验验证目标差异基因对通路的抑制/促进。
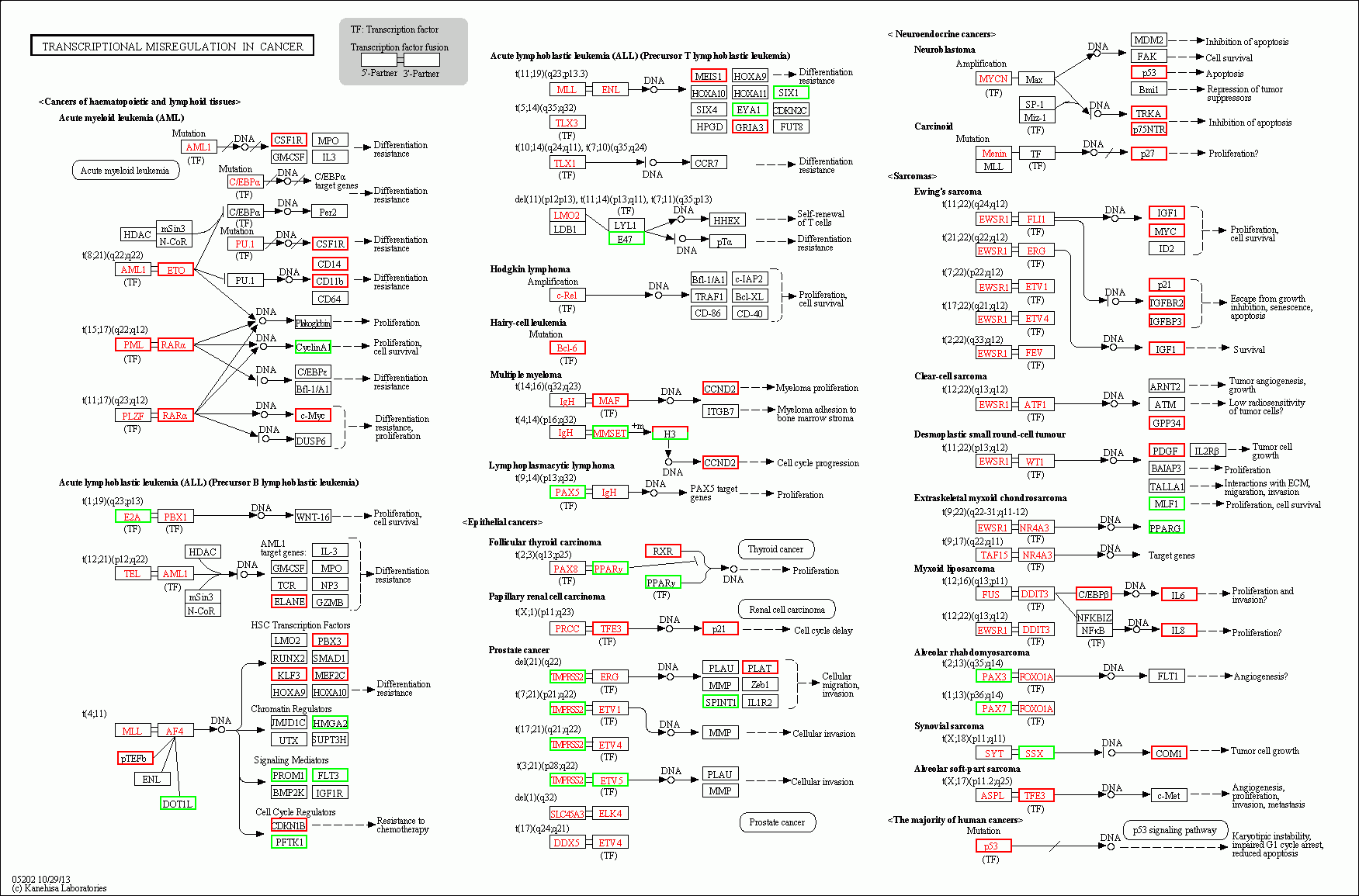
通路map图中的实线和虚线各代表什么?
实线箭头:①一步生化反应;②正向调控;③分子相互作用。
虚线箭头:①间接反应;②不确定的生化反应。
KO富集气泡图中的rich factor指的什么?代表的含义?怎么计算?
Rich factor为该代谢路径下差异基因数目与所有注释到该路径基因数目的比值,数值越大表示富集程度越大。可以用clusterprofiler R包去计算得到。
Q8:转录组归一化的含义?发挥的作用?
A8:归一化指的是在比较不同样本转录组表达量的时候,需要量化成一个标准,然后将raw counts同时除以目标基因的外显子长度之和(也就是目标基因转录本长度)和总的有效比对的read总数,这就是归一化处理。
我们在比较不同样本的转录本表达量时,是基于归一化处理的数据文件进行的。
Q9:如何理解测序随机性图(即reads在参考基因组上的分布)?随机性好坏的标准是什么?
A9:随机性是测序质量的一个判定因素,目前尚没有标准来评估随机性的好坏。但通常来说,如果测序随机较好,reads会较均匀地分布在参考序列上。
Q10:原始数据都包含了哪些内容?原始数据可以用什么软件打开查看?
A10:测序得到的原始图像数据经base calling转化为序列数据,我们称之为Raw data,结果以FASTQ文件格式存储,包含reads的序列以及碱基的测序质量。在FASTQ格式文件中每个read由四行描述,如下:
@A80GVTABXX:4:1:2587:1979#ACAGTGAT/1
NTTTGATATGTGTGAGGACGTCTGCAGCGTCACCTTTATCGGCCATGGT
+
BTTMKZXUUUdddddddddddddddddddddddddddadddddd^WYYU
每个序列共有4行,第1行和第3行是序列名称(有的fq文件为了节省存储空间会省略第三行“+”后面的序列名称),由测序仪产生;第2行是序列;第4行是序列的测序质量,每个字符对应第2行每个碱基,第四行每个字符对应的ASCII值减去64,即为该碱基的测序质量值,比如c对应的ASCII值为99,那么其对应的碱基质量值是35。
可以用记事本或者notepad ++打开;但是有时会出现FASTQ文件打不开的情况,这是什么原因呢?
有可能是因为文件太大,在这种情况下我们可以尝试用pycharm读取文件,或者抽取一部分数据再打开。
Q11:将Clean Data比对到核糖体是为了看什么?
A11:受样品质量和物种的影响,实验方法去核糖体的效率可能不太稳定,而核糖体的污染会影响后续的分析,因此首先使用短 reads 比对工具 bowtie 将 High quality clean reads 比对到核糖体数据库,最多允许 5 个错配,去除比对上核糖体的 reads,将保留下来的数据用于后续的分析。如果比对到核糖体上的reads占比很低,则说明数据质量比较好,可以用于后续分析。
今天对转录组的常见问题解答整理先分享到这里,如果您有其他疑惑,欢迎随时联系我们~