Metagenomics(元基因组学,或者宏基因组学),是由 Handelman等1998年最先提出的一种直接对微生物群体中包含的全部基因组信息进行研究的手段。宏基因组测序是对特定环境样品中的微生物群落基因组进行高通量测序,以分析微生物群体基因组成多样性与丰度,探求微生物与环境以及宿主之间的关系,发掘和研究新的、具有特定功能的基因。它规避了对样品中的微生物进行分离培养的过程,提供了一种对不可分离培养的微生物进行研究的途径,更真实的反应样本中微生物组成、互作情况、系统进化等,同时在分子水平对其代谢通路、基因功能进行研究。
应用方向
1. 健康和疾病人群细菌分类鉴定、属菌丰度统计、差异菌群Biomarker 筛选
2. 挖掘健康和疾病人群微生物群落中的功能基因等作为Biomarker
3. 疾病与微生物菌群、菌落中的功能基因关系的研究
实验方案
测序平台与方式:Illumina平台 PE150
数据量:大于6G clean data
技术优势
1. 单次测序产生至少6G数据量,有利于发现特异物种信息,深度挖掘基因资源;
2. 无需分离培养微生物,对微生物群落多样性和功能基因等宏观特征进行研究,能更有效、准确的反应出微生物的真实状态;
3. 相较于16S rDNA测序,宏基因组不仅可以对细菌分类进行分析,还能进行基因和功能层面的深入研究;
4. 16S rDNA测序一般注释到属水平,而宏基因组能鉴定到种水平甚至菌株水平,并且16S测序只扩增了高变区的片段,宏基因组是将微生物基因组打断并拼接组装成较长的序列,可以发现更多低丰度物种信息,构建不同物种之间的代谢网络途径,因此在鉴定物种过程中具有更高的优势;
技术流程

图1 宏基因组测序实验流程
数据分析
3.1数据质控
3.2 Metagenome组装
3.3 基因预测
3.4 物种注释
3.5 功能注释
3.6物种和功能分析
3.7抗性基因分析:获得抗性基因丰度分布情况以及这些抗性基因的物种归属和抗性机制
3.8其他高级分析:如CCA/RDA分析,肠型分析,拷贝数变异(CNV)分析,CAG/MLG分析,病原与宿主互作数据库(PHI)注释,分泌蛋白预测, Ⅲ型分泌系统效应蛋白预测,细菌致病菌毒力因子(VFDB)注释,转移元件分析(MGE)等;同时,结合环境因子、病理指标或特殊表型进行深入关联研究,能够为进一步深入研究和利用样品的物种和功能提供理论依据。
送样建议
样本来源:人、大鼠和小鼠(其它样本类型请咨询)
人粪便样本:>2g/样;动物粪便>0.5g/样
肠道内容物样本:200mg/样
肠道组织样本:>1g/样品,块状物需要黄豆样大小 1~2 粒/样
运输条件:干冰运送
结果展示
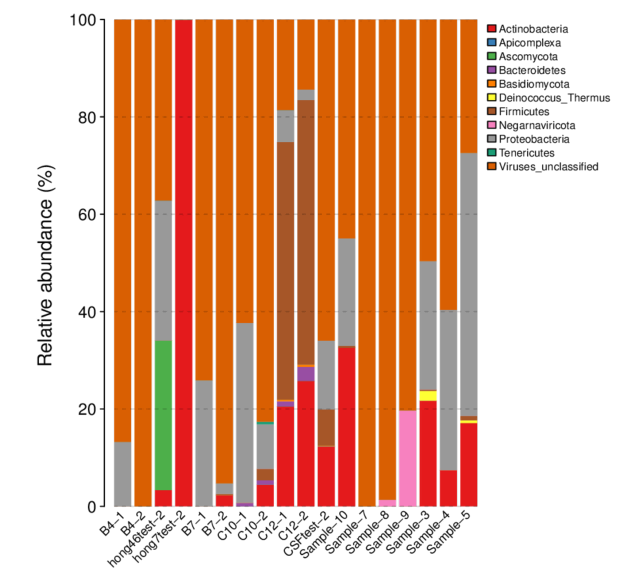
图2 各样品Phylum水平的物种相对丰度柱形图
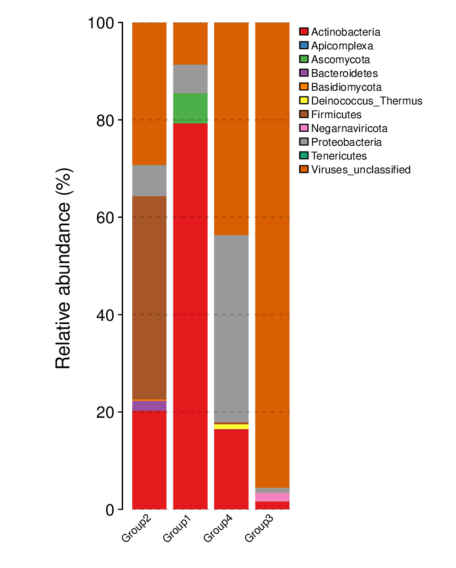
图3 各组Phylum水平的物种相对丰度柱形图
(从不同分类层级的相对丰度表出发,并将丰度 < 0.5% 的物种设置为 Others,绘制出各样品对应的物种注释结果在不同分类层级上的相对丰度柱形图)
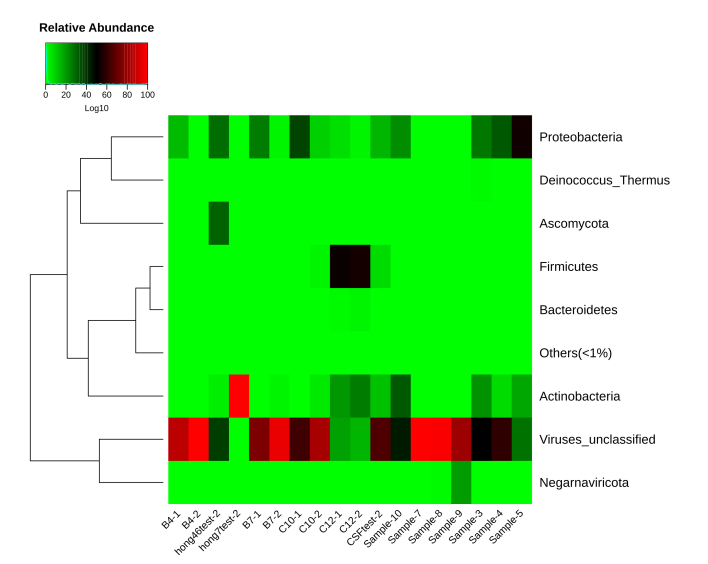
图4 各样品Phylum水平的物种相对丰度热图
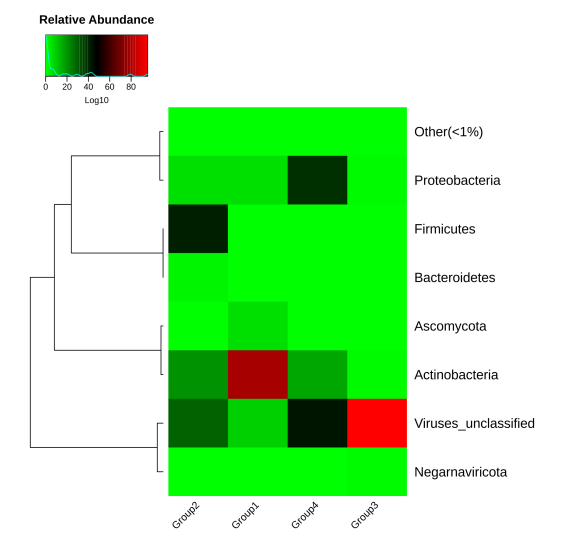
图5 各组Phylum水平的物种相对丰度热图
(从不同分类层级的相对丰度表出发,每个样品中的丰度信息绘制热图,并从物种层面进行聚类,便于结果展示和信息发现,从而找出样品中聚集较多的物种)
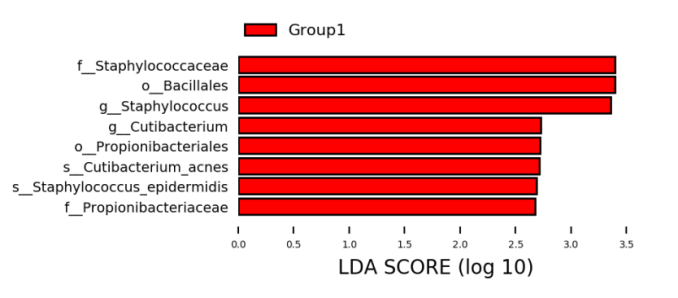
图6 基于LDA score筛选的biomarker

图7 特征物种的柱状环形图

图8 KEGG注释基因数目统计图
案例分析 :利用宏基因组新一代测序技术洞察肺结核患者独特的肺菌群图谱
Insights into the Unique Lung Microbiota Profile of Pulmonary Tuberculosis Patients Using Metagenomic Next-Generation Sequencing[1]

研究方案:为探究与肺结核相关的微生物群分布情况,并研究抗结核治疗期间的纵向变化,研究者采集8 名健康人(HCG)、12 名未治疗的肺结核患者(UTG)、15 名已治疗的肺结核患者(TTG)、11 名治愈的肺结核患者(CTG)和 7 名肺癌患者(LCG)的支气管肺泡灌洗液(BALF)样本,并在健康对照组中采集咽拭子。利用metagenomic sequencing分析比较指定组中微生物群的差异,对结核病患者的肺部微生物群进行了表征,并评估抗结核治疗对肺微生物群的影响。
研究结果:本研究对所有队列数据进行了主坐标(CAP)分析,发现在属和种水平上,咽拭子和 BALF 样本之间的微生物群组成存在明显分离。为了研究上呼吸道和下呼吸道微生物群的差异,研究者比较了健康对照的咽拭子和 BALF 间的微生物群,发现咽拭子中的ACE指数和Chao1指数在种和属水平上均远高于BALF,香农指数在种水平上显着高于BALF,但在属水平上无显着差异,而辛普森指数在种或属水平上这两组样本之间没有显着差异。主坐标分析 (PCoA) 显示,在种和属水平上,BALF 簇与咽拭子簇明显分离,表明上呼吸道和下呼吸道之间的微生物群组成不同。进一步研究者分析了咽拭子和 BALF 间相对丰度top 30的物种,发现咽拭子和BALF中top30的细菌分别占细菌总数的61.58%和84.03%,其中,Klebsiella pneumoniae、Staphylococcus aureus、Pasteurella multocida是三组中的优势物种。为了研究长期抗结核治疗后治愈的结核病患者的肺微生物群的变化机制,研究者比较了HCG和CTG之间的微生物组成,发现2组的多样性指数没有显著差异,通过LEfSe分析(LDA SCORE>2.0)也并未发现两组之间有存在差异的物种,结果表明治愈的结核病患者表现出与健康人相似的肺微生物群结构和组成。进一步研究发现CTG比HCG显示出更多的多样性和丰富的ARGs,并在 cooccurrence network中显示CTG中细菌和ARGs的连接要比HCG中的复杂。结果表明,肺结核患者区别于健康个体和肺癌患者表现出独特的微生物组成,抗结核病治疗增加了肺微生物群的α多样性并影响了β多样性,加速了ARGs的富集。通过对肺部微生物群进行分析比较有助于更好地了解肺结核的发病机制。

图9 所有样本中的菌群情况
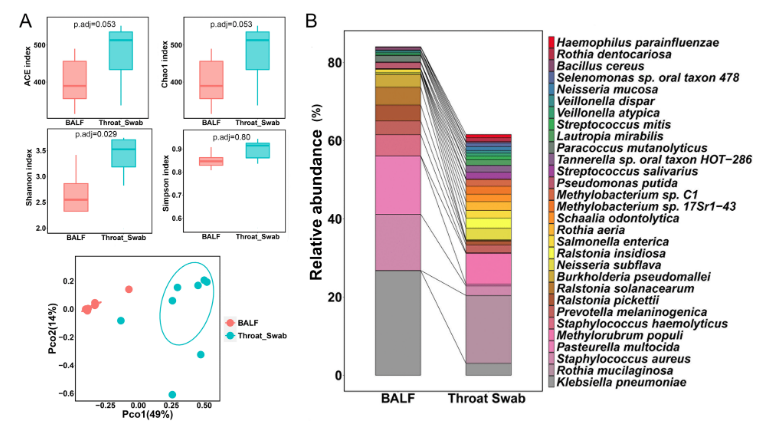
图10 来自健康对照组的咽拭子和BALF样本之间的菌群分布有显著差异
参考文献:Xiao G, Cai Z, Guo Q, et al. Insights into the Unique Lung Microbiota Profile of Pulmonary Tuberculosis Patients Using Metagenomic Next-Generation Sequencing[J]. Microbiology spectrum, 2022, 10(1): e01901-21.